


Did Project Warp Speed Warp the Regulators?
Evidence of fraud within the regulatory system?

Your support is very much appreciated. Please consider taking out a paid subscription to support independent journalism.

Thanks to one of my readers for making me aware of this information. It was posted on Market-Ticker yesterday, so for the full document please click the link.
The document “Did Pfizer Perform Adequate Safety Testing for its Covid-19 mRNA Vaccine in Preclinical Studies? Evidence of Scientific and Regulatory Fraud” was written by Sasha Latypova. Sasha’s background is in pharmaceutical research and development. Pharmaceutical companies often outsource clinical trials to contractors and she used to be one of those contractors, collecting data. She has done work for over 60 different pharmaceutical companies, including Pfizer and Johnson and Johnson with years of experience reading documents from clinical and preclinical studies.
Sasha looks a some of the recently released Pfizer documents and highlights “its deficiencies, omissions and gaps that were clearly visible, yet never questioned by the regulators or other health authorities.” She concludes that these omissions and the dishonesty cannot be attributed to incompetence by the manufacturers and regulators.
Specifically, she identified seven findings which led her to her conclusion of scientific and regulatory fraud.
1.Pfizer’s program did not include a comprehensive end-to-end test of all components of the final approved product. The studies included in the approval package were for a variety of versions of the product with no comparability assessments, thus no comprehensive assessment of product safety can be made.
mRNA vaccines were novel platforms comprising of a “payload + delivery vehicle” structure. This meant Pfizer were required to assess the safety of ALL components separately, as well as the final assembled version.
However, Sasha found that multiple versions of each component were used without separate tests for each new version. Pfizer’s Phase I clinical trial used BNT162b1, which was never used because b2 was eventually selected and new versions were added without new testing.
She identified 18 non-clinical studies of which only 9 “directly related to the licensed product and to only some of the components of the final product”.
Even more concerning, in September 2021, the FDA issued draft guidance “Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial”, however this included an exception stating it did not apply to “vaccines intended to prevent infectious diseases”. Sasha thinks this unprecedented deviation from the regulatory standard could be to cover their tracks but also notes that it shouldn’t apply anyway because the vaccine doesn’t actually prevent disease.
2. The toxicity of the vaccine’s mRNA active ingredient was never studied!
As I have written about before, one of the studies looked at whether the vaccine stayed at the injection site or travelled around the body. However, Sasha points out, Pfizer didn’t test the actual vaccine (encoding the spike protein), they used a surrogate.
By doing this, the public and medical professionals are fooled into believing that the surrogate (lipid nanoparticles) are the “product” without any care for what the payload is or does. She uses the analogy of trying to compare a truck loaded with food and a truck loaded with explosives and saying they are the same thing because they are delivered by a truck. The fact that the FDA did not find this objectionable, she finds shocking.
3. Pfizer claimed absence of potential for “vaccine-elicited disease enhancement” based on studies of an animal species that does not get sick from Sars-Cov-2.
Again, I have written about disease enhancement before and will do again shortly due to what I am reading in the latest documents. Whilst the FDA were aware of the risk, Pfizer claimed there was no evidence of vaccine-elicited disease enhancement based on one animal study comprising of 9 monkeys, three of which were given a placebo. None of the monkeys got clinically ill with Pfizer concluding because of this, this was an infection model, not a COVID-19 disease model. Why did the FDA not pick up on this?
4. CDC, FDA and Pfizer lied about “vaccine staying in the injection site”
Although the rat study was again based on a surrogate, not the spike protein version itself, it clearly showed major accumulations in organs around the body. This graph shows the concentrations when the study was halted.
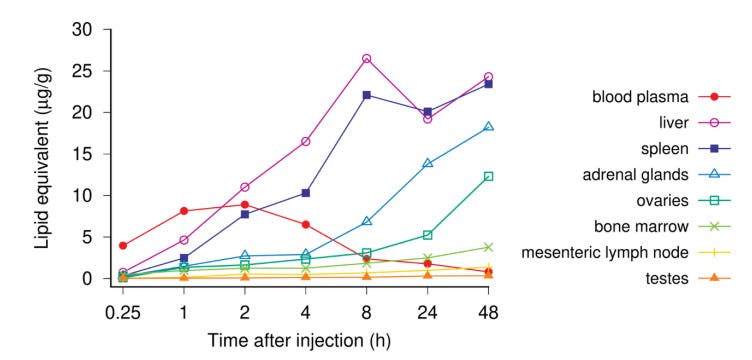
“No follow-on studies elucidating the complete time course of distribution, time to maximum concentration, maximum concentrations observed, and time to clearance were performed or planned. No estimates of the therapeutic safety margins were provided”.
Sasha notes that the European Medicines Agency noticed this when they made the following comments. “No traditional pharmacokinetic or biodistribution studies have been performed with the vaccine candidate BNT162b2….Biodistribution: Several literature reports indicate that LNP-formulated RNAs can distribute rather nonspecifically to several organs such as spleen, heart, kidney, lung, and brain. In line with this, results from the newly transmitted study 185350, indicate a broader biodistribution pattern”. Why did the FDA not spot this?
5. Pfizer skipped major categories of safety testing altogether.
Pfizer decided not to study:
Secondary Pharmacodynamics (assessment of new drugs for pharmacological activities other than the desired therapeutic target).
Safety Pharmacology (how the drug acts on the body and where it goes, how long it stays there and how it is eliminated)
Pharmacodynamic Drug Interactions (potential harmful effects when interacting with existing medicines)
Genotoxicity (evaluate the risk of possible DNA cellular damage)
Carcinogenicity (evaluate the risk of cancer formation)
She asks “what rationale was used to waive these entire categories of pharmacological safety testing?”
6. Pfizer used dishonest and self-serving interpretation of regulatory guidelines to avoid routine safety testing.
Pfizer claimed the studies above were not necessary due to guidelines produced for vaccine development by the WHO in 2005.
“Pfizer’s product was only arbitrarily reclassified as a vaccine in 2020. Prior to that it was categorized as a gene therapy, so back in 2005 when the WHO guidelines were written, it would not have been regarded as a vaccine. Furthermore, 2005 recommendations from WHO did not anticipate gene therapy platforms. Additionally, it is the responsibility of the FDA and other regulatory bodies worldwide to regulate the authorization and licensing of medical products. WHO does not have this authority as it is only an advisory and coordination non-governmental body.”
Why did the FDA not assert its authority over the WHO guidelines?
7. Both FDA and Pfizer knew about major toxicities associated with gene therapy class of medicines, and therefore cannot claim lack of anticipatory knowledge of these risks. This points to intentional fraud and collusion between Pfizer and the regulators to push this untested dangerous product on the market.
Sasha list numerous FDA guidelines which exist for studying cellular and gene therapy products. These guideline clearly anticipate many risks including known risks such as:
Multi-organ failure and death
Potential for tumors/cancer development
Late onset T-cell leukemia
Potential for prolonged uncontrollable activity after single administration
Immunogenicity as a risk (autoimmunity)
Uncontrolled expression of genes
Migration of product to undesired organ systems
Possibility of shedding: excretion/secretion of viral particles that could be transmitted to other individuals
Studies in healthy volunteers are not generally advised due to potential severe toxicities
“The guidance also states that the risks associated with the gene therapy class may be entirely novel and cannot be derived from prior history of other drug classes. In other words, this class is uniquely risky and requires an extensive and rigorous safety testing program. It should be noted that prior to 2020, all gene therapy derived products were being developed for extremely severe, often terminal illnesses like terminal cancer and Huntington’s disease. They could not be even tested in healthy people, much less prescribed to every human on the planet as a prophylactic treatment, and much less forced on every human being regardless of consent”.
Sasha says Pfizer chose to disregard these guidelines and the FDA let them get away with it. “Pfizer’s dishonest interpretation of guidelines and cherry-picking of the applicable regulations resulted in brazen disregard for all routine safety assessments”.
She also notes that one paragraph from the latest guidance says studies may not be required if the products are derivatives of “approved safe products”. Does this give Pfizer and Modern future “outs” on safety testing whilst making it more difficult for other manufacturers?
She concludes by saying “The mandate of the FDA as the industry regulator requires the agency to question and check such reckless disregard for safety testing. An honest regulator would have questioned the assertion by the manufacturer that major categories of safety studies were not applicable to their product. There is no question of incompetence. The agency is staffed with qualified and experienced pharmacology and toxicology professionals. At this point, with millions of adverse event reports accumulating rapidly in every public health database, neither the FDA, NIH, CDC, Pfizer nor other manufacturers can claim ignorance of these issues. The question of fraud and wilful negligence by both the manufacturers and the regulators must be raised”.
For a longer explanation of some of these issues by Sasha herself, please click here to watch a related video.
trump rushed through these so-called vaccines with new technology. not smart.
Operation Warp speed will come to be remembered as the worse political decision ever.
The link is broken already. You can find the article at https://home.solari.com/review-of-pfizers-non-clinical-program-by-sasha-latypova/